

Background:
Several specific therapies for MDS are now available. Molecular genetics plays a key role, as some therapies target distinct mutations and others are particularly effective for molecularly defined subgroups (e.g. SF3B1). In parallel, broad mutation studies suggest a continuous progression from clonal cytopenia of undetermined significance (CCUS) to MDS. Since panel sequencing is available for routine laboratory workflows, we evaluated its potential for diagnosis of CCUS and extended the question to possible (future) therapeutic decisions.
Aim:
(1) Identify the frequency of clonality (mutations) in a real life MDS vs. cytopenia cohort; (2) subdivide CCUS with a low and high risk of developing MDS; (3) identify the subset of patients qualifying for personalized treatments
Patients and Methods:
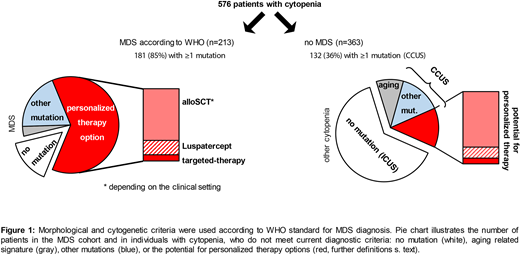
We performed morphology and cytogenetic analysis according to current WHO gold standard algorithms in 576 patients (F:221, M:355; median age: 72 [17-94]) with cytopenia and clinical conditions requiring bone marrow biopsy to exclude or diagnose MDS. Individuals with other possible explanations (e.g. lymphoma, PNH) had been excluded upfront by combining morphology and flow cytometry. DNA was isolated from bone marrow; sequencing was performed on NovaSeq after NextFlex library preparation (Illumina, ILMN, San Diego, CA) and hybrid capture of a 41 gene panel (IDT Inc. Coralville, IA). Data was analyzed with Pisces and Pindel (for FLT3-ITD) (BaseSpace, ILMN) using a minimum sensitivity of 3%. Genetic variants were classified by combining databases and in silico predictions.
Results:
We found 499 mutations in 181/213 (85%) patients with diagnosis of MDS, and 259 mutations in 132/363 (36%) other cytopenic patients (p <0.01). Cases with mutations are by definition CCUS and without mutation ICUS (idiopathic cytopenia of undetermined significance). CCUS and MDS patients were significantly older than ICUS (CCUS: 74, MDS: 75 vs. ICUS: 66 years, p <0.02, each). MDS patients had significantly more mutations per case than CCUS (average: 2.8 vs. 2.0, p<0.01). By contrast, the typical aging pattern (DNMT3A, TET2 and ASXL1 mutations [DTA] with VAF <10%) as the only molecular genetic aberration was significantly more frequent in CCUS than in MDS with mutations: 25% vs. 4% (33/132 vs. 7/181; p<0.01).
The International Working Group for the Prognosis of MDS - IWG-PM - (Malcovati, et al. Blood 2020) suggested SF3B1 mutations to define the first subgroup of patients by molecular genetics. Using their criteria, 12% of our MDS (26/213) patients fall into this novel category of SF3B1-mutated MDS. Due to the high risk of developing MDS, the IWG-PM suggests to use the mutation as presumptive evidence of MDS even without definitive morphological features. In our CCUS cohort the same genetic SF3B1 constellation was significantly less frequent, however 5% of individuals (7/132, p=0.03) qualified for MDS diagnoses by molecular genetics. In addition, CCUS patients show other MDS typical mutations: e.g. 4% (5/132) had RUNX1 mutations and 30% (39/132) had splicing factor mutations other than SF3B1. Malcovati et. al. (Blood 2017) defined a "highly specific pattern" of mutations for patients with a 20% risk of developing a myeloid malignancy per year. This pattern was frequent in MDS (71%, 151/213), but was detected in 55% of CCUS (72/132), which corresponds to 20% of cytopenia patients without a definitive MDS diagnosis (72/363).
Next, we subdivided the cohorts according to their potential treatment options (Figure 1). (A) SF3B1 mutations would suggest use of Luspatercept. (B) According to Bejar et al. (ASH 2015), 95/213 (45%) of our MDS and 37/132 (28%) of our CCUS have a high-risk genetic profile. Individuals with high-risk mutations could be considered for alloSCT depending on progression of disease or clinical settings. (C) A potential for targeted therapies (mutations in CSNK1A1, FLT3 [ITD, TKD], IDH1, IDH2, KRAS, NRAS) was found in 6% (12/213) of the remaining MDS and 3% (4/132) of the CCUS cohort.
Conclusions:
We suggest that molecular genetic markers should be recognized as presumptive evidence of MDS and allow the diagnoses to be based on three cornerstones: morphology, cytogenetics and molecular genetics. In two thirds of MDS and one third of CCUS patients mutations can be identified that allow personalized treatment based on respective molecular genetics even independent of other currently used diagnostic labels.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal